
Antibody deficiency most frequently results in recurrent and severe sinopulmonary infections with encapsulated bacterial strains (eg, Streptococcus pneumoniae, Haemophilus influenzae). Children commonly present with recurrent otitis media, sinusitis, and pneumonia. Adults present similarly, although otitis media is less common. Viral infections of the respiratory tract also occur with greater frequency and severity in these patients. Clinical presentation is discussed in more detail elsewhere.
The age of the patient can help narrow the differential diagnosis:
- The most common antibody defects that present in infancy are transient hypogammaglobulinemia of infancy, selective antibody deficiency (after the age of two years) and selective IgA
- The most common antibody deficiencies in young children are selective antibody deficiency, selective IgA deficiency, IgG subclass deficiency, and early-onset common variable
- The most common disorders that present in adulthood include common variable immunodeficiency, selective IgA deficiency, IgG subclass deficiency, and selective antibody
Measurement of antibody levels. Measurement of immunoglobulin G (IgG), IgA, and IgM is useful in all cases of suspected antibody deficiency.
There are several methods for determining serum Ig levels, and laboratories use different systems. Therefore, it is critical that age-adjusted normal reference ranges are provided. Hypogammaglobulinemia is defined as an IgG less than two standard deviations from normal, and agammaglobulinemia is usually considered when IgG is <100 mg/dL. Panhypogammaglobulinemia (ie, low levels of IgA, IgG, and IgM) is a hallmark of most forms of severe combined immune deficiency. In other combined immune deficiencies, as well as in several predominantly humoral immune deficiencies, there are characteristic alterations in the profile of immunoglobulin (Ig) isotypes that may aid in diagnosis (eg, selective IgA deficiency, selective IgM deficiency, and hyper IgM immunodeficiencies).
In addition to IgG, IgA, and IgM, measurement of IgG subclasses and IgE may be useful:
- Measurement of IgG subclasses may be valuable in children older than one year of age and in adults, particularly if the IgG level is borderline low and/or there is a weak or absent antibody response to
- Measurement of IgE is helpful in patients with recurrent sinopulmonary infections, as an elevation is consistent with underlying allergic A very elevated IgE (ie, >2000 IU/mL) in a patient with recurrent bacterial infections and dermatitis would raise suspicion for hyperimmunoglobulin E syndrome.
Measurement of serum IgD is not useful for the diagnosis of immune deficiency.
Measurement of antibody function. Antibody function can be assessed by measuring antibody titers (usually IgG) to specific organisms (aka specific antibody) in response to intentional immunization or natural infection. Antibody function is critical for determining the integrity of humoral immunity for two reasons:
- Clinically significant impairment in antibody function can be present even when serum antibody levels are
- Normal responses to immunization (ie, normal antibody function) can occur with subnormal levels of total IgG or of IgG This pattern is typically seen with secondary causes of hypogammaglobulinemia, such as that caused by medications, protein loss, or severe malnutrition.
Antibody function is assessed by examining the patient’s response to the two general types of antigens; protein antigens and polysaccharide antigens. Routine vaccinations provide examples of both types:
- Measurement of antibody titers to tetanus, diphtheria, Hemophilus influenzae B, and protein- conjugated pneumococcal vaccines (eg, Prevnar) are used to assess response to protein Antibody titers to other vaccines (eg, hepatitis A and hepatitis B, measles, others) can also be used.
- Measurement of antibody titers to multiple serotypes in pneumococcal polysaccharide vaccines are used to assess response to polysaccharide This evaluation is useful in adults and children older than two years. This response is particularly important in making a diagnosis of selective antibody deficiency.
Isohemagglutinins are antibodies generated in response to polysaccharides of gut flora and which cross-react with A or B blood group erythrocyte They generally appear in the blood by six months of age in individuals who have blood types other than AB. However, vaccine
responsiveness is generally considered to be a more reliable indicator of intact humoral immune function. Isohemagglutinin testing is reviewed in more detail separately.
Assessing vaccine responses is reviewed in detail separately.
Measurement of immune globulin loss. Low levels of immunoglobulins are occasionally due to loss of immune globulin into the GI tract, urine, lung, skin, or peritoneal fluid during dialysis in patients with certain chronic diseases. A method of evaluating the rate of IgG loss is reviewed separately.
Defects in cellular immunity
Specific cellular immunity is mediated by T cells, and defects affecting these T cells underlie the most severe immune deficiencies. However, because antibody production by B cells requires intact T cell function, most T cell defects lead to combined (cellular and humoral) immune deficiency.
An evaluation of cellular immunity is appropriate for patients with severe viral and/or bacterial illnesses or opportunistic infections. The disorders that can impair cellular immunity are different in various age groups:
- In children younger than one year of age, primary immunodeficiencies are the most common cause of impaired cellular immunity, although perinatal cytomegalovirus and other herpes virus infections can cause transient or persistent cellular
- In older children and adults, the major causes are HIV infection and iatrogenic immune suppression due to therapy for autoimmune disease, malignancy, or Occasionally, mild forms of primary combined immune deficiency or DiGeorge syndrome escape diagnosis until adolescence or adulthood.
The most profound combined immunodeficiencies are classified under the heading “severe combined immunodeficiency” or SCID. SCID disorders usually present in infancy, while less severe combined immunodeficiencies present in children and occasionally, in adolescents or adults.
Complete blood count with differential and blood smear. The complete blood count (CBC) with differential and blood smear provides valuable information about the cellular immune system. Normal lymphocyte counts in infants are much higher than in older children and adults. In many primary immunodeficiency disorders, cell populations are initially normal, and then decline over time. Thus, normal results in the past cannot be relied upon as a reflection of the current state.
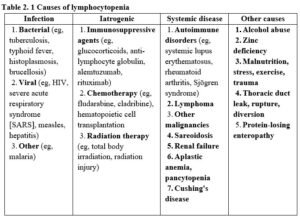
In patients suspected of having a defect in cellular immunity, the CBC establishes the presence or absence of lymphopenia and any associated gross hematologic abnormalities, some of which may greatly assist in diagnosis. A single finding of lymphopenia should be interpreted with caution, since transient lymphopenia is frequently found in a variety of common infectious illnesses. However, significant lymphopenia that does not rapidly correct should not be ignored, since lymphopenia may be the first indication of cellular immunodeficiency or another serious disease (eg, lymphoma) (table 2. 1). In rare situations, a normal lymphocyte count can be seen in the presence of a severe immunodeficiency. If the clinical presentation is highly suggestive of an underlying disorder (eg, a patient with Pneumocystis pneumonia and invasive candida), then lymphocyte subsets should be evaluated with flow cytometry even if the total lymphocyte count is normal.
Evaluation of lymphopenia. Lymphopenia can be caused by an array of disorders. Flow cytometry to evaluate lymphocyte populations should be performed in all patients suspected of cellular immunodeficiency with any significant infection and a total lymphocyte count <1500 cells/microliter (<2500 cells/microliter in infants). In the absence of other indicators of immune dysfunction, there should at the very least be subsequent measurement of the lymphocyte count to document normalization. Persistent lymphopenia requires further investigation of immune function.
Cutaneous delayed-type hypersensitivity. Cutaneous delayed-type hypersensitivity (DTH) is the classic in vivo test of cellular immunity. This test measures the recall response to an intradermal injection
of an antigen to which an individual has already been exposed over a period of time For that reason skin testing is usually not of much value under age two.
A positive response to intracutaneous antigen injection requires uptake and processing of antigen by antigen presenting cells, their interaction with CD4+ helper T cells, cytokine production by T cells, and subsequent recruitment and activation of monocytes and macrophages. Thus, skin testing is a sensitive indicator of intact cellular immunity, but negative results must be interpreted with caution, because an impairment in any step of the response pathway will lead to a negative response. In addition, the DTH response is unreliable in children under one year of age (even with prior antigen exposure), and is often suppressed during viral and bacterial infection (even with live attenuated vaccines as in the combined measles, mumps, and rubella vaccine). DTH skin reactivity is also suppressed by antiinflammatory drugs (glucocorticoids) and other immunosuppressants (cyclosporine tacrolimus mycophenolic acid).
Method. Delayed hypersensitivity skin responses can be assessed using an intradermal injection of Candida antigen. This is the only commercially-available reagent intended for use in DTH testing. A 0.1 mL dose of undiluted Candin® is injected intradermally in the volar surface of the forearm. The response is measured 48 to 72 hours after injection. Induration of more than 5 mm is considered positive in all cases. In children, induration of more than 2 mm is sometimes accepted as positive. Erythema alone does not indicate a positive reaction.
In one author’s center (ERS), a panel of four reagents is used for DTH testing: Candin® (as above), Tetanus toxoid (10 flocculation units/mL, Sanofi Pasteur, diluted to 1.5 LF/mL), PPD (5 TU/mL, Sanofi Pasteur), and Trichophyton (1:500 w/v, Allermed Laboratories).
The main advantages of DTH testing are its ease and economy; it is a useful screening test in many instances of suspected cellular immune deficiency. A negative DTH should be followed either with repeated testing (often following a tetanus toxoid booster immunization if tetanus was among the testing reagents), or with measurement of lymphocyte populations by flow cytometry, combined with in vitro
assays of T cell function, as discussed below. In vitro assays (particularly mitogen stimulation) are less sensitive to interference during intercurrent illnesses or by drugs.
Flow cytometry. Flow cytometry uses monoclonal antibodies to identify and quantitate hematopoietic cells that have specific antigens termed cluster designations (CDs). The table lists the fundamental set of markers commonly used and the lymphocyte populations they define (table 2.2). A typical panel of markers used to identify the major subset of lymphocytes includes CD3, CD4, CD8, C19/20, CD16/56/57. The nature and derivation of the nomenclature of the markers are discussed elsewhere.

A CBC with differential should be performed on a blood specimen obtained at the time of the flow cytometric analysis, or the cytometer itself be used to determine the lymphocyte number. This analysis permits the calculation of the absolute numbers of each lymphocyte subset. It is possible for the percentage of a particular subset to be abnormal while the total number of cells is within the normal range, and vice-versa. An absolute deficiency, rather than a relative (percentage) deficiency, is of much greater clinical significance.
Flow cytometry is invaluable in the assessment of lymphocyte subpopulations in patients with opportunistic infections or severe or persistent lymphopenia. Standard flow cytometry analysis will be abnormal in almost all cases of severe combined immune deficiency (SCID), and in many instances of other combined immunodeficiencies. The analysis of lymphocyte subsets may be diagnostic for various forms of lymphoma as well. The use of flow cytometry in the diagnosis of specific immunodeficiencies is reviewed in more detail separately.
Abnormalities in immunodeficiency. The table summarizes anticipated alterations in the representations of various lymphocyte populations in several immune deficiency diseases. Specific disorders are reviewed separately.
While certain immune defects are associated with characteristic patterns of lymphocyte subsets, lymphocyte populations may appear to be entirely normal even with clinical evidence of significant immune dysfunction. Conversely, as with total lymphocyte numbers, lymphocyte subsets may be profoundly altered by common infectious illnesses and other factors. Thus, for the purpose of diagnosis, flow cytometry data must be considered in conjunction with functional tests of the immune system.
Decreased numbers or absent natural killer CD16/56 cells (≤100 cells/ul), particularly in the presence of recurrent infection with herpes viruses, should warrant further studies for natural killer cell deficiencies, including cytotoxicity studies.
Advanced tests. Many advanced tests are available to study specific cellular immune defects. These tests are best ordered and interpreted by an immunology expert. Advanced tests include:
- Advanced flow cytometry using monoclonal antibodies specific for activated cells, regulatory T cells, naïve or memory cells, or various stages of B cell development are of value in further characterizing many antibody deficiencies (eg common variable immunodeficiency) or cellular immunodeficiencies (eg immune dysregulation, polyendocrinopathy, enteropathy, X-linked or IPEX syndrome). Flow cytometry can be used in the definitive diagnosis of several genetic immunodeficiencies by assessing the absence or abnormal expression of a specific protein (eg, X- linked agammaglobulinemia, Wiskott-Aldrich syndrome).
- Analysis of T cell receptor excision circles (TRECs) provides an assessment of thymic function by measuring recent thymic emigrant This is also used as a screening test for newborn SCID on newborn blood samples.
- Cytotoxicity assays measure the functional capacity of cytotoxic T cells or natural killer T cell cytotoxicity is a researeh tool since the target cells must share HLA antigens with the patient. One such method utilizes antigenic peptides attached to specific MHC molecules labeled with a fluorochrome as targets. Binding of the cytotoxic cell to the peptide is measured by flow cytometry.
- Cytokine measurements on cells, serum, or tissue
- Mutation analysis to identify mutated genes for many T cell or combined
- Chromosome analysis is of value in the diagnosis of DiGeorge syndrome.
- HLA typing is used to identify chimerism in newborns with SCID or to identify an appropriate stem cell
- CD3/T cell receptor analysis and function.
In vitro studies of T cell function. In vitro studies of T cell function measure peripheral blood T cell proliferation in response to several different types of stimuli:
- Mitogens (such as the plant lectins phytohemagglutinin, concanavalin A, pokeweed mitogen, anti-CD3)
- Specific antigens (such as tetanus and diphtheria toxoids or C. albicans antigens)
- Allogeneic lymphocytes (ie, mixed lymphocyte culture)
These studies may not be possible in patients with profound lymphopenia. In parallel with (or prior to) studies of T cell function, flow cytometric measurement of peripheral blood lymphocyte subpopulations should be performed.
In T cell function studies, the patient’s purified peripheral blood mononucleareells (lymphocytes and monocytes) are incubated in sterile media with the test substance(s) or cells for three to six days. A control tube with cells and media alone is also incubated. During the last 24 hours of culture, tritiated thymidine is added to the cultures. Dividing lymphocytes incorporate the thymidine into their DNA. The extent of proliferation is determined by measuring the radioactivity taken up by cells. The test is interpreted as negative, partial, or normal by comparing patient results with normal control lymphocytes assayed simultaneously and with the normal ranges for controls in the laboratory.
Many laboratories also report results as a stimulation index (SI). This is the ratio of radioactivity (as counts per minute, or CPM) with stimulation over the CPM in the control tube without stimulation (background).
Suppression of T cell responses to mitogens and antigens may be seen with significant nutritional deficiencies, moderate to severe concurrent illnesses, or with administration of immunosuppressive drugs. Thus, these tests should be performed when the patient is relatively well and not receiving glucocorticoids or other immunosuppressive medications, whenever possible. T cell responses appear to normalize rapidly (ie, within a day or two) after glucocorticoids are discontinued.
Response to mitogens. Most mitogens require functional antigen presenting cells (ie, monocytes and B cells) for T cell stimulation, although the mechanisms of antigen processing and presentation are bypassed. Mitogens are powerful stimulants for T cells, and responses may be assessed in patients of any age, even newborns, regardless of immunization status.
Normal reference ranges are derived from studies on the cells of healthy adults. Newborns generally have higher mitogen responses compared to adults. Depending upon the mitogens and the laboratory, the range of counts per minute (CPM) reported for normal controls is approximately 50,000 to 300,000 and SI between 10 and 200. Phytohemagglutinin (PHA) is most commonly used. Results for patients in comparison to controls are roughly interpreted as normal (>50 percent of control), low (25 to 50 percent), very low (10 to 25 percent), or absent (<10 percent). Results expressed as SI vary widely between laboratories. In general, a SI <10 may be considered as no response.
Some researehers have also used in vitro stimulation with the OKT3 monoclonal antibody as a measure of T cell function. OKT3 binds to the epsilon chain of the T cell CD3 complex and stimulates T cells (in the presence of accessory or antigen presenting cells) in a manner analogous to T cell mitogens. Cell response to mitogen or OKT3 is generally similar, although they may sometimes give different patterns in the context of distinct immune defects. There are no published comparisons in a large group of normal individuals or immune deficient patients. Most clinical reference laboratories use one or more mitogens and one or more antigens, but do not routinely use OKT3.
Diminished or absent proliferation signals a serious derangement of T cell function. Mitogen responses will be severely depressed (usually well below the control fifth percentile) in the vast majority of patients with profound lymphopenia (eg, complete DiGeorge syndrome), severe combined immune deficiency (SCID), or advanced AIDS. By comparison, the response may be partial or normal in milder syndromes of combined deficiency (eg, Wiskott-Aldrich syndrome), in mainly humoral deficiency, or in those with infections (especially CMV and other herpes viruses).
Response to specific antigens. Specific antigen tests are more sensitive than mitogen assays as indicators of defects in T cell function, since the mechanisms of activation are more complex than those for mitogens. These tests require antigen processing and presentation by antigen presenting cells, although T cell recruitment of effector cells (eg, in vivo DTH) is not necessary. As with DTH, in vitro specific antigen proliferation tests are not useful in infants or other patients who have not completed a primary series of immunizations.
Tetanus and diphtheria toxoids and monilia (Candida) are antigens frequently used for this assay. Since only a small number of cells respond, the cultures must be maintained longer, and incorporation of radioactivity is lower than for mitogen stimulation. Results expressed as CPM are often reported from 5000 to 50,000; results as SI generally range from 43 to 50. Because of the very large variation in the frequency of antigen-specific T cells in the blood of healthy controls, the SI is often most important for interpretation. In general, a SI for specific antigen >43 is considered adequate.
In many cases of combined immune deficiency, responses to specific antigens are diminished or even absent while mitogen responses remain intact. In secondary immune deficiency, specific antigen responses will also generally be suppressed more easily than mitogen responses. However, these
generalizations are not always true. As with DTH testing, booster immunization with tetanus followed by a repeat in vitro proliferation assay may detect a significant response.
Another way to assess function of T cells is to measure cytokine release following antigen exposure. An example of this type of test that has become widely used in the diagnosis of latent tuberculosis infection is the interferon release assay, which can be used instead of tuberculin testing.
Neutrophil defects (defects in phagocyte function). Neutrophil defects result in a range of illness from mild recurrent skin infections to overwhelming, fatal systemic infection. Affected patients are more susceptible to bacterial and fungal infections, but have a normal resistance to viral infections. Most are diagnosed in infancy due to the severity of the infection or the unusual presentation of the organism, but some escape diagnosis until adulthood.
Primary phagocytic deficiencies characteristically lead to recurrent and severe fungal (eg, Candida and Aspergillus) and bacterial (eg, Staphylococcus aureus, Pseudomonas aeruginosa, Nocardia asteroides, Salmonella typhi) infections. Response to nontuberculous mycobacteria may also be abnormal, particularly in patients with chronic granulomatous disease. The most common sites of infection are the respiratory tract and skin. Tissue and organ abscesses also occur. Other frequent manifestations include abnormal wound healing, dermatitis/eczema, stomatitis, and delayed umbilical cord detachment. Many patients have growth failure.
Phagocytic disorders can be caused by extrinsic or intrinsic defects. Extrinsic defects include opsonic abnormalities secondary to deficiencies of antibody and complement factors, suppression of granulocyte production, and leukocyte autoantibodies or isoantibodies that decrease the number of circulating neutrophils. Intrinsic disorders of granulocytes may be divided into defects in granulocyte killing ability and defects in chemotaxis (cell movement).
Evaluation of neutrophil numbers. The initial test for evaluation of phagocyte disorders in the CBC with differential:
- Leukocytosis raises the possibility of a leukocyte adhesion defect (ie, a defect in chemotaxis), and flow cytometry is indicated next to assess for the presence of specific adhesion
- Severe neutropenia can be seen with congenital agranulocytosis or cyclic neutropenia.
- If neutrophil numbers are normal and the patient’s history is consistent with a neutrophil disorder, then an evaluation of neutrophil function is
Evaluation of neutrophil function. Disorders of neutrophil function include chronic granulomatous disease (CGD), Chediak-Higashi syndrome, neutrophil-specific granule deficiency, leukocyte adhesion defects, and hyper IgE syndrome. Initial tests include:
- Neutrophil oxidative burst is best measured by a flow cytometric assay of dihydrorhodamine (DHR). This test is quantitative and is useful in the identification of heterozygote carriers of This procedure is widely available in reference laboratories and has generally replaced the nitroblue tetrazolium test (NBT test). However, the NBT test can be used for screening purposes.
- An IgE level, which is often elevated in hyper IgE
- Examination of a peripheral blood smear to detect:
- The giant azurophilic granules in neutrophils, eosinophils, and other granulocytes, which are diagnostic of Chediak-Higashi
- Neutrophils that lack granules and are often bilobed, which are found in neutrophil-specific granule
- Advanced mutation analysis and flow cytometric analysis for many neutrophil disorders are also
- A chemotaxis assay can be done to identify a cell movemen
Complement defects
Complement disorders may be inherited or acquired. Screening for a classical complement pathway defect is indicated in patients with any of the following:
- Recurrent, unexplained pyogenic infections in whom the white blood count and immunoglobulin levels and specific antibody responses are normal
- Recurrent Neisserial infections at any age
- Multiple family members who have experienced Neisserial infections
In addition, it is reasonable to evaluate the complement system in any patient with systemic lupus erythematosus (SLE). However, this is particularly relevant in those with familial lupus or subacute cutaneous lupus, in whom C1q or C2 deficiency should be excluded.
The initial screening test for complement defects is a total hemolytic complement assay (CH50), which assesses classical pathway function and is widely available. If the CH50 is significantly reduced or zero, then the levels of individual complement components are measured. Low levels of multiple components, particularly low C3 and C4, suggest complement consumption, rather than a primary complement deficiency. If the CH50 is normal and a complement defect is still suspected, the AH50, a screening test for alternative pathway defects, may be obtained. This test is largely performed in specialized laboratories. Alternative complement deficiencies include properdin and factor D deficiencies, both of which are rare. Inherited and acquired defects in complement components are reviewed in more detail elsewhere.
If a patient has frequent infections suggestive of a complement deficiency, a mannose-binding lectin defect should be excluded.
Defects and deficiencies of C1 esterase inhibitor are associated with hereditary angioedema, although these disorders are not immunodeficiencies because these patients do not have increased susceptibility to infection.
Innate immune defects
Defects in innate immune mechanisms are relatively rare disorders that should be suspected in patients in which other immunodeficiencies have been excluded. Many are associated with specific infections (eg, herpes or EBV infections in NK cell defects, atypical mycobacterial infection in IL-12/23- IFN-γ pathway defects, neonatal herpes simplex encephalitis in the TLR-3 signaling pathway and other severe infections, often associated with poor inflammatory response).
Natural killer cell defects — NK cell defect can be primary or secondary. Primary NK deficiency should be suspected in patients with low NK cells and/or recurrent severe herpes virus infections. NK defects are also noted in patients with X-linked lymphoproliferative disorders and Hemophagocytic lymphohistiocytosis, Chediak-Higashi syndrome.
Defects of IL-12/23-IFN-gamma pathways — Patients with invasive infections caused by low virulence Mycobacterial and Salmonella species may have defects of the genetic components of the IL- 12/23-IFN-γ pathways including the IFN-γ receptor 1 gene (IFNGR1) or the IFN-γ receptor 2 gene (IFNGR2), the IL-12 receptor β1 gene (IL12RB1), the IL12B gene, and the STAT1 gene. Special tests involving western blotting or flow cytometry can pinpoint the exact molecular defects.
Toll-like receptor pathway defects — Pattern recognition receptors (PRRs) are receptors specific for moleculareomponents of microorganisms widely expressed on phagocytic cells (neutrophils, macrophages, and monocytes and several other cells). Defects in PRRs can cause increased susceptibility to infections that is most severe in infancy, before the adaptive (ie, humoral and cellular) immune system has developed. The frequency and severity of infections generally lessen as the patient matures, in contrast to most other types of immunodeficiency.
The Toll-like receptors (TLRs) are one group of PRRs that, after ligation with specific microorganisms, initiate a series of steps that eventually activate the nucleus to initiate cytokine synthesis.
Several genetic disorders of the signaling pathways have been identified, including an IL-1 receptor- associated kinase 4 defect (IRAK4), a myeloid differentiation primary response gene 88 defect (MYD88), a nuclear factor kappa B essential modulator defect (NEMO), a TLR3 receptor defect, and an UNC-93B defect. Screening tests to evaluate these defects are available, including a flow cytometric assay.


